Agénésie des valves pulmonaires

Radiographie thoracique:
Hypertrophie des hiles pulmonaires et de l’arc moyen gauche, emphysème obstructif ou présence d’atélectasies, cardiomégalie (au dépend de l’arc inférieur droit et gauche essentiellement), absence d’hypervascularisation pulmonaire périphérique[1].
Définition et épidémiologie
C’est une anomalie extrêmement rare, surtout de manière isolée.
Il s’agit d’une absence de valvules pulmonaires
L’anomalie est facilement détectée en échographie anténatale, par la visualisation d’une dilatation des artères pulmonaires ainsi que dans la majorité des cas une CIV.[3]
Embryologie
Dans la majorité des cas, cette malformation est associée à une anomalie génétique, la délétion 22q11 ou syndrome de Di George.
L’origine embryologique n’est pas connue à ce jour, et l’association quasi systématique à une agénésie du canal artériel a fait proposer différentes hypothèses dont aucune n’a réellement fait sa preuve.
Description anatomique et variantes
La région valvulaire présente dans cette pathologie un tissu valvulaire atrophique et dysmorphique ainsi qu’une anomalie de l’anneau.
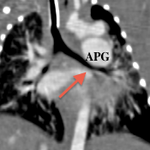
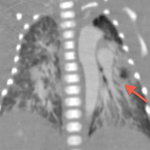
Il en résulte une régurgitation et plus ou moins une sténose valvulaire pulmonaire responsable d’une dilatation anévrysmale du tronc de l’artère pulmonaire ainsi de que de ces branches principales venant comprimer l’arbre trachéobronchique. [2]
Cette compression des voies aériennes est un problème thérapeutique majeur, la chirurgie ne le résolvant pas toujours.
Dans plus de 90% des cas, cette anomalie est associée à une tétralogie de Fallot. Au départ d’ailleurs, on décrivait ces formes comme une variante de Fallot, mais leur pronostic est propre et bien plus sombre.[2,3]
L’agénésie du canal artériel est quasi systématique, associé à une CIV.
De rares cas décrits présentent un canal artériel présent avec un trajet inhabituel et ont un septum ventriculaire intact.[4]
D’autres cardiopathies complexes ont été reportées en association, comme une atrésie tricuspidienne, un VDDI.
Traitement
Il est uniquement chirurgical[5], et consiste à une réparation complète en cas de tétralogie de Fallot, associée à une plastie de réduction des artères pulmonaires, selon les équipes jusqu’aussi loin que possible.
D’autres ont proposé une réimplantation du tronc de l’artère pulmonaire en avant de l’aorte pour retirer tout risque de compression ; il a également été proposé la mise en place de stent sur les voies aériennes.
Il est également réalisé une reconstruction de la valve pulmonaire, souvent monocusp de nos jours.
L’age moyen de la chirurgie est de 0,8 ans dans une étude récente.
Evolution à long terme
Les mortalités néonatale et post-opératoire ne sont pas négligeables. On compte environ 20% de mortalité postopératoire[3].
La survie après chirurgie est estimée à 82% à un an et 79% à 5, 10 et 15 ans par Norgaard[5].
La dépendance ventilatoire en pré-opératoire semble être le principal facteur pronostique d’une issue fatale.
Imagerie
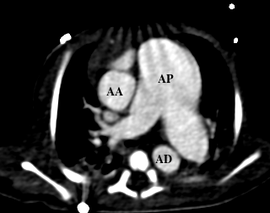
Que ce soit en pré- ou en post-opératoire, l’imagerie en coupes a une place primordiale dans cette pathologie en permettant une étude précise de l’arbre trachéobronchique, des niveaux de compression, de la morphologie des artères pulmonaires et de leurs tailles ainsi que de la présence ou non d’un canal artériel[6,7,8].
Le scanner peut avoir l’avantage en pré-opératoire d’être d’exécution rapide chez ces enfants souvent en dépendance ventilatoire et de permettre une analyse de la naissance des coronaires, une tétralogie de Fallot étant souvent associée.
L’IRM permet également l’analyse fonctionnel de la valve pulmonaire grâce à des séquences SSFP ciné oblique centrées sur la voie d’éjection droite et de mesurer les débits de régurgitation tant en préopératoire qu’en postopératoire.
Les séquences axiales morphologiques T1 black Blood permettent de détailler l’anatomie des artères pulmonaires.
Des séquences coronales apprécieront mieux l’arbre trachéobronchique et l’artère pulmonaire droite alors que des séquences sagittales apprécient l’artère pulmonaire gauche.
Il faut également s’attacher à rechercher les anomalies associées : présence d’une CIV, mesure de la fonction ventriculaire droite et gauche, des voies de dérivation systémicopulmonaires (surtout en cas d’agénésie d’une branche pulmonaire).
GALERIE D'IMAGES:

Bibliographie
- Macartney FJ, Miller GAH. Congenital absence of the pulmonary valve. British heart J.1970; 32: 483-490
- Kastler B, Clair C, Livolsi A, Narboux Y, Delabrousse E, Sarliève P. Malformations des artères pulmonaires. Cardiopathies cyanogènes : aspects IRM.EMC Radiodiagnostic, 2002 ; [ 32-015-F-70]
- Wertaschnigg D, Jaeggi M, Chitayat D, Shannon P, Ryan G, Thompson M, Yoo SJ, Jaeggi E.Prenatal diagnosis and outcome of absent pulmonary valve syndrome: Contemporary single center experience and review of the litterature. Ultrasound Obstet Gynecol 2012;doi: 10.1002/uog.11193.
- Podzimkova J, Hickey MS, Slavik Z, Leanage R, Chen Chan K. Absent pulmonary valve syndrome with intact ventricular septum: Role of ductus arteriosus revisited. Intern J Cardiol 1997; 61: 109-112
- Nørgaard MA, Alphonso N, Newcomb AE, Brizard CP, Cochrane AD. Absent pulmonary valve syndrome. Surgical and clinical outcome with long-term follow-up. European Journal of Cardio-thoracic Surgery .2006; 29: 682—6–75
- Taragin BH, Berdon WE, Printz B. MRI assessment of bronchial airways compression in absent pulmonary valve syndrome and review of the syndrome. Pediatr Radiol 2006;36: 71-75
- Lambert V, Sigal-Cinqualbre A, Belli E, Planché C, Roussin R, Serraf A, Bruniaux J, Angel C, Paul JF. Preoperative and postoperative evaluation of airways compression in pediatri patients with 3D MSCT: Effect on surgical management. J Thorac Cardiovasc Surg 2005; 129: 1111-8
- Frank H, Salzer U, Popow C, Stiglbauer R, Wolleneck G, Imhof H. Magnetic resonance imaging of absent pulmonary valve syndrome. Pediatr Cardiol 1996; 17: 35-39
Auteurs
Clio Sorensen
clio_13007@hotmail.com
Interne des hopitaux d'Amiens
Dr Guillaume Gorincour
Service d'imagerie pédiatrique de l'hopital de la Timone enfants