Hétérotaxie
Définition et épidémiologie
Le mot hétérotaxie vient du grecque « hétéros » qui signifie diffèrent et « taxis » qui signifie arrangement [1]. Ce terme est donc employé dès lors qu’il existe une anomalie du situs, c’est à dire dès que les organes ne sont pas disposés de la manière dont ils le sont habituellement (situs solitus). Ce syndrome n’est pas restreint au système cardiovasculaire, mais intéresse l’ensemble des organes (cœur, poumon, foie, rate).
On définit par isomérisme, dans le contexte des malformations cardiaques, des structures paires, de cotés opposés, à droite et à gauche du rachis, qui sont morphologiquement identiques, formant une symétrie, une image en miroir.
Isomérisme droit défini donc un cœur où les deux oreillettes auront une morphologie droite. A l'opposé, isomérisme gauche définit un cœur où les deux oreillettes auront une morphologie gauche.
La fréquence de ces anomalies est difficile à estimer avec précision car certaines sont asymptomatiques (essentiellement des formes d’isomérisme gauche), l’hétérotaxie représente 0,8% des cardiopathies congénitales [8,9].
Embryologie
Initialement, l’embryon est parfaitement symétrique, puis au cours de l’organogénèse, les structures vont devenir droite ou gauche. Cette latéralisation survient très tôt au cours du développement. Certains gènes permettent ainsi la différentiation de la partie droite de l’oreillette primitive en oreillette droite et inversement. Les cellules savent déjà ce qu’elles vont devenir en quelque sorte. Toutes anomalies génétiques bloquant cette signalisation cellulaire aboutit à des anomalies du situs.
Il n’existe pas un seul gène impliqué, mais il s’agit plutôt d’une origine plurifactorielle.
Clinique
Isomérisme droit
L’histoire naturelle est sombre, avec une survie à un an proche de 0%, étant donné la fréquence des anomalies cardiaques complexes, notamment des retours veineux pulmonaires anormaux totaux, ainsi que du risque de choc septique étant donné l’asplénie.
Les manifestations cliniques sont donc dominées par une détresse respiratoire, une cyanose ou un sepsis sévère, souvent en période néonatale [1].
Isomérisme gauche
La survie à 1 an sans intervention est d’environ 50%, meilleur que dans l’isomérisme droit. L’apparition de signes d’insuffisance cardiaque en période néonatale est le principal signe, moins souvent la cyanose [2].
Description et variantes anatomiques
Isomérisme droit

L’anomalie princeps est la présence de deux oreillettes morphologiquement droites ainsi que deux poumons trilobés. Les bronches principales sont toutes deux épiartérielles. Il s’y associe une asplénie, d’où l’autre dénomination de cette anomalie, syndrome d’asplénie ou syndrome d’Ivemark.
Fréquemment, la veine cave inferieure est à gauche de l’axe rachidien et l’aorte ipsilatérale à celle-ci [2].
Le foie est médian, l’estomac en position indéterminée.
Ce type est plus fréquent chez le garçon.
Les anomalies cardiaques sont fréquentes :
- Anomalies du retour veineux pulmonaire, surtout totales
- Canal atrioventriculaire
- Double veine cave supérieure
- Transpositions des gros vaisseaux
- Cœur univentriculaire, ventricule droit à double issue
- Sténose et atrésie pulmonaire [3]
Les autres anomalies associées à retenir sont les infections sévères dues à l’asplénie et les malrotations intestinales.
Isomérisme gauche

Plus fréquent chez la fille.
Dans ce cas, les deux oreillettes sont de morphologie gauche avec deux poumons bilobés et des bronches souches hypartérielles.
Il s’y associe une polysplénie, d’où l’autre dénomination de cette anomalie sous le terme de syndrome de polysplénie.
Fréquemment, il existe une interruption suprarénale de la veine cave inférieure avec continuation azygos et hémiazygos. Classiquement, l’aorte est localisée à gauche du rachis.
Le foie est médian, l’estomac en position variable.
Les anomalies cardiaques sont moins fréquentes que dans l'isomérisme droit, retrouvées dans pres de 50% des cas [1]:
- Anomalies du retour veineux pulmonaires (partielles essentiellement)
- Canal atrioventriculaire
- Double veine cave supérieure
- Obstruction de la voie de sortie gauche, coarctation aortique
- Sténose ou atrésie pulmonaire
- Transpositions des gros vaisseaux, ventricule droit à double issue
- Communication interauriculaire de tout type, communication interventriculaire [4]
Les anomalies associées à retenir sont l’atrésie des voies biliaires, les blocs auriculo-ventriculaires et les malrotations intestinales.
Traitement
Le traitement chirurgical, lorsqu’il est possible, est très variable. Il dépend des anomalies cardiaques rencontrées, des anomalies veineuses systémiques ou pulmonaires [1,3,4,5,6].
Il existe un ventricule unique fonctionnel dans la majorité des cas. Le traitement sera uniquement palliatif, par la réalisation de l'intervention de Fontan modifié, ou en cas d’interruption de la veine cave inférieure, par l’intervention de Kawasaki [1].
Certaines équipes parlent également de l’intérêt de la transplantation cardiaque dans les cas les plus complexes.
Evolution à long terme
Malgré les progrès de la chirurgie, ce syndrome reste l’une des malformations les plus complexes, avec un pronostic réservé, notamment en cas d’isomérisme droit.
La survie moyenne est estimée à 70% tous types confondus [1].
Dans le cas d’isomérisme droit, la mortalité moyenne est de 70%, avec 25% de patients non opérés, et un pronostic plus réservé pour ceux opérés dans la période néonatale. La survie moyenne est de 70% à 1mois, 49% à 1 an et 35% à 5 ans [3].
Dans un article plus récent, la survie moyenne à 5 ans est de 81,4% à 5 ans.[4]
Les facteurs de risque indépendants de mortalité sont : l’absence de sténose pulmonaire, la présence d’une anomalie majeure de la jonction atrioventriculaire, un obstacle au retour veineux [3].
Dans le cas de l’isomérisme gauche, en cas de réparation biventriculaire, la survie moyenne est de 80% à 1 an, 71% à 5 ans, 66% à 10 ans et 48% à 15 ans[6]. Dans les cas d’orientation vers un cœur univentriculaire fonctionnelle, la survie 73% à 1 an, 61 à 5 ans, 53% à 10 ans et 48% à 15 ans [6].
Une autre étude ne montre pas de difference significative en terme de survie selon que la réparation soit de type bi ou univentriculaire et estime une probabilité desurvie à 15 ans de 50%[7].
Les facteurs de risque de mortalité identifiés sont surtout les troubles de la conduction cardiaque et les anomalies extracardiaques, 40% des cas (atrésie des voies biliaires ou malrotation intestinale)[6,7]
Imagerie
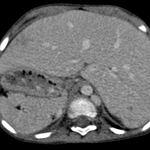
Etant donné la complexité de ce syndrome et les variantes nombreuses, l’imagerie en coupes a toute sa place dans son évaluation.
Que ce soit l’IRM ou le scanner, ils permettent une étude précise tant de l’anatomie cardiaque que des poumons et de l’abdomen et sont supérieurs en terme diagnostic pour l’analyse des anomalies veineuses systémiques ou pulmonaires [8,9].
Ces techniques permettent une vue d’ensemble de l’anatomie avec précision ce que ne permet ni l’échographie, ni la cathétérisme.
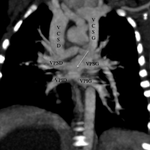
Dans ce sens, le radiologue doit s’efforcer de définir :
- la morphologie des oreillettes
- la morphologie bronchique
- la présence ou non d’une interruption de la veine cave inférieure
- la présence ou non de retour veineux pulmonaires anormaux
- décrire une éventuelle veine cave supérieure gauche
- une communication interauriculaire ou ventriculaire
- un canal atrioventriculaire
- l’aspect des artères pulmonaires et leur taille
- l’aspect de l’aorte, la recherche d’une coarctation aortique
- une anomalie conotroncale : un aspect de ventricule droit à double issue, une tétralogie de Fallot, un tronc artériel commun, une transposition des gros vaisseaux (bien que déjà diagnostiqué par l’échocardiographie)
- dans le cas de l’IRM, la fonction ventriculaire, surtout en cas de ventricule unique fonctionnel, en vue d’une intervention de dérivation cavopulmonaire supérieure ou Fontan[9]
- au niveau abdominale : l’aspect du foie, de la rate, la position de l’aorte et de la vésicule biliaire et une éventuelle malrotation intestinale qui sera mieux vue au scanner qu’à l’IRM.





Bibliographie
- Kim S-J. Heterotaxy syndrome. Korean Circ J 2011;41:227-232
- Applegate KE., Goske MJ., Pierce G., Murphy D. Situs revisited : imaging of the heterotaxy syndrome. Radiographics.1999 ; 19 : 837-852
- Hashmi A., Abu-Sulaiman R., Mccrindle BW., Smallhorn JF., WIilliams WG, Freedom RM. Management and Outcomes of Right Atrial Isomerism : A 26-Year Experience J Am Coll Cardiol 1998; 31:1120–6
- Ota N., Fujimoto Y., Murata M., Tosaka Y., Ide Y., Tachi M., Ito H, Sugimoto A., Sakamoto K. Improving Outcomes of the Surgical Management of Right Atrial Isomerism. Ann Thorac Surg 2012; 93:832–9
- Gilljam T., McCrindle BW., Smallhorn JF., Williams WG., Freedom RM. Outcomes of Left Atrial Isomerism Over a 28-Year Period at a Single Institution. J Am Coll Cardiol. 2000; 36:908–16
- Eronen MP., Aittoma ̈ki KAU. , Kajantie EO., Sairanen HI. Outcome of Left Atrial Isomerism at a Single Institution Pediatr Cardiol. 2012 ; 33:596–600
- Chen SJ, Li YW, Wang JK, Wu MH, Chiu IS, Chang CI, Hsieh SC, Su CT, Hsu JC, Lue HC. Usefulness of electron beam computed tomography in children with heterotaxy syndrome.Am J Cardiol. 1998 Jan 15;81(2):188-94.
- Geva T., Vick GW., Wendt RE., Rokey R. Role of spin echo and cine magnetic resonance imaging in presurgical planning of heterotaxy syndrome. Comparison with echocardiography and catheterization. Circulation. 1994; 90:348-356
Auteurs
Clio Sorensen
clio_13007@hotmail.com
Interne des hopitaux d'Amiens
Dr Guillaume Gorincour
Service d'imagerie pédiatrique de l'hopital de la Timone enfants