Anomalies de la septation ventriculaire
Radio tx : cardiomégalie avec bombement de l’arc moyen gauche et hypervascularisation pulmonaire.
Définition et épidémiologie
Les anomalies de la septation ventriculaire (CIV) sont l’anomalie cardiaque congénitale la plus fréquente (30 à 40%[1,2]des cardiopathies congénitales). La meilleure détection de celles-ci par l’échographie a fait augmenter le nombre de CIV détectées. Dans une étude récente, la prévalence était de 53/1000 naissances et 90% de ces défects septaux se ferment spontanément dans les 10 premiers mois de vie.[3]
Embryologie
La connaissance du devellopement du septum inter-ventriculaire permet une meilleure approche de ces defects. Celui resulte du devellopement et de la fusion de differentes structures embryologiques : les bourgeons endocardiques du conotroncus, les bourgeons endorcardiques atrioventriculaires et le septum interventriculaire primitif, musculaire. On comprend ainsi qu’une anomalie du développement ou de la fusion d’une de ces parties aboutit à une CIV.
Anatomie déscriptive et variantes décrites
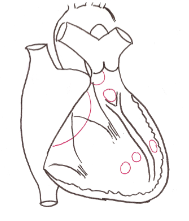
La classification la plus employée est celle d’Anderson[4] mais néanmoins aucune classification n’a fait l’unanimité.
On distingue néanmoins :
- Les CIV musculaires : situées sur le septum musculaire, elles se ferment de manière spontanée la plupart du temps.
- Les CIV membraneuses : juxta-artérielles, infundibulaires ousupracristagalis au contact des valves semilunaires.
- Les CIV péri-membraneuses : elles s’étendent selon trois directions : la voie d’admission, le septum infundibulaire, le septum musculaire.
A cette classification purement anatomique, s’oppose une classification fonctionnelle[5], définit par les données échographiques : A COMPLETER
Les CIV sont soit isolées soit « incluses » dans une cardiopathie complexe comme une tétralogie de Fallot, un VDDI, une transpositions des gros vaisseaux ou une anomalie chromosomique telle que la trisomie 18, 13 ou 21(CIV d’admission+++).
D’autres anomalies associées sont à connaître : canal artériel persistant, CIA, VCSG se drainant dans le sinus coronaire, anomalies valvulaires.
Dans le cadre de CIV isolées, on décrit une association fréquente des CIV juxta-artérielles ou péri-membraneuses à une insuffisance aortique, du fait de l’absence de soutien sous-valvulaire et d’un prolapsus de la sigmoïde aortique antérodroite : c’est le syndrome de Laubry et Pezzi.[1]
CIV avec sténose pulmonaire (type 4 de classification hémodynamique) : cette anomalie permet de protéger l’arbre artériel pulmonaire.
CIV et bloc auriculoventriculaire : c’est l’apanage des formes péri-membraneuses où se trouve le tissu de conduction (dans leur portion postéro-inférieure). Le risque est surtout post-opératoire.[2]
Clinique et évolution à long terme
La fermeture spontanée est plus ou moins fréquente selon le type de CIV, surtout dans les cinq premières années [6], mais on observe des fermetures spontanées jusqu’à l’adolescence. Il faut retenir qu’une insuffisance aortique peut apparaître dans 10% des cas.[7]
Les CIV musculaires ont tendance à la fermeture spontanée sauf celles proche du septum d’admission.
Les CIV périmembraneuses peuvent se fermer spontanément surtout si elles sont proches de la valve septale de la tricuspide et si elles sont de petite taille.
Quant aux CIV juxta-artérielles, elles se ferment très rarement spontanément.
L’évolution naturelle d’une CIV non restrictive (c’est à dire assez grande pour ne pas gener un shunt important) peut se faire vers l’insuffisance cardiaque gauche, une hypertension artérielle pulmonaire, rare de nos jours grâce à une prise en charge adaptée.
L’avènement de la chirurgie a profondément transformé la survie des patients avec des CIV non restrictives, avec une mortalité opératoire de 1% et une survie à long terme de plus de 90%. Une étude récente montre même un taux de survie de 99,5% à 2 ans.[8]
Les principales complications de la chirurgie sont des troubles conductifs, rythmiques et hémodynamiques.
Traitement
Le traitement chirurgical est le plus employé. Il consiste à la fermeture de la CIV par un patch en tissu synthétique. La voie trans-atriale ou trans-pulmonaire est préférée pour éviter les cicatrices ventriculaires pourvoyeuses de troubles du rythme.
En cas d’insuffisance aortique associée, une plastie aortique sera également réalisée.
Le cerclage de l’artère pulmonaire n’est plus réalisée de nos jours devant les excellents résultats de la chirurgie correctrice.[2,8]
La traitement par cathétérisme avec mise en place d’un AmplatzerÒ peut également être réalisé, notamment pour les CIV musculaires multiples.[1,2]
Imagerie
Scanner
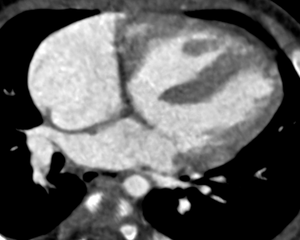
Le scanner n’a pas d’intérêt dans le bilan des CIV. Néanmoins, elles sont largement détectables sur des scanners cardiaques.[9] Le radiologue doit connaître celles-ci et savoir les décrire de manière aussi précise que possible lorsqu’il en détecte une sur un examen. Il est également important d’étudier les artères pulmonaires, leur taille et la présence éventuelle de signes d’hypertension pulmonaire.
IRM
En préopératoire ou préinterventionel :
L’IRM est peu réalisée en pratique pour ce genre de pathologie, sauf si elle rentre dans le cadre d’une pathologie plus complexe ( tétralogie de Fallot, APSO…). L’échographie est en effet largement suffisante tant pour le diagnostic que pour le suivi. Néanmoins dans certains cas de CIV haute, ou de patient peu échogène, l’IRM a montré sa capacité à détecter de manière précise la localisation, la taille et les bords des CIV ainsi que sur leur retentissement hémodynamique.
La mesure du rapport Qp/Qs est un bon indicateur mais peut être mis en défaut en présence d’une fuite aortique ou pulmonaire. Ce ratio peut être obtenu par la mesure des fractions d’éjection ventriculaire gauche et droite ou par des séquences d’encodage de flux au niveau de la racine aortique et pulmonaire.
Il faut alors préférer la mesure directe du flux trans-orificielle, en obtenant un plan de coupe dans le plan de la CIV dit « in face », permettant une mesure directe du shunt.[10]
Une récente étude montre également qu’une simple mesure du Qp permet d’estimer, par une équation les résistances vasculaires pulmonaires avec une bonne sensibilité. Cela pourrait à l’avenir permettre de diminuer les procédures invasives.[11]
En post intervention :
L’IRM n’a pas forcement d’indication mais peut montrer un shunt résiduel.

Bibliographie
- Penny DJ, Vick GW 3rd. Ventricular septal defect The Lancet 2011; 377: 1103–12
- Chantepie A. Communications interventriculaires.EMC Cardiologie.2005 ; 11-940-C-30
- Roguin N, Du ZD, Barak M, Nasser N, Hershkowitz S, Milgram E. High Prevalence of Muscular Ventricular Septal Defect in Neonates JACC. 1995 ; 26(6):1545-8
- Soto B, Becker AE, Moulaert AJ, Lie JT, Anderson RH. Clasification of ventricular septal defects. Heart J 1980; 43: 332-343
- Nadas AS. Diagnosis and treatment : management of ventricular septal defect. Pediatrics. 1964 Aug; 34:271-3.
- Onat T, Ahunbay G, Batmaz G, Celebi A. The Natural Course of Isolated Ventricular Septal Defect During Adolescence. Pediatr Cardiol 1998; 19:230–234,
- Turner SW, Hornung T, Hunter S.Closure of ventricular septal defects: a study of factors influencing spontaneous and surgical closure. Cardiol Young 2002 Jul;12(4):357-63.
- Scully BB, Morales DL, Zafar F, McKenzie ED, Fraser CD Jr, Heinle JS.Brandi. Current Expectations for Surgical Repair of Isolated Ventricular Septal Defects. Ann Thorac Surg 2010; 89:544–51
- Rajiah P, Kanne JP. Computed tomography of septal defects. J Cardiovasc Comput Tomogr. 2010 Jul-Aug; 4(4):231-45.
- Rajiah P, Kanne JP. Cardiac MRI: Part 1, Cardiovascular Shunts. AJR 2011; 197:W603–W620
- Bell A, Beerbaum P, Greil G, Hegde S, Toschke AM, Schaeffter T, Razavi R. Noninvasive Assessment of Pulmonary Artery Flow and Resistance by Cardiac Magnetic Resonance in Congenital Heart Diseases With Unrestricted Left-to-Right Shunt. JACC Cardiovasc Imaging. 2009 Nov;2(11):1285-91
Auteurs
Clio Sorensen
clio_13007@hotmail.com
Interne des hopitaux d'Amiens
Dr Guillaume Gorincour
Service d'imagerie pédiatrique de l'hopital de la Timone enfants